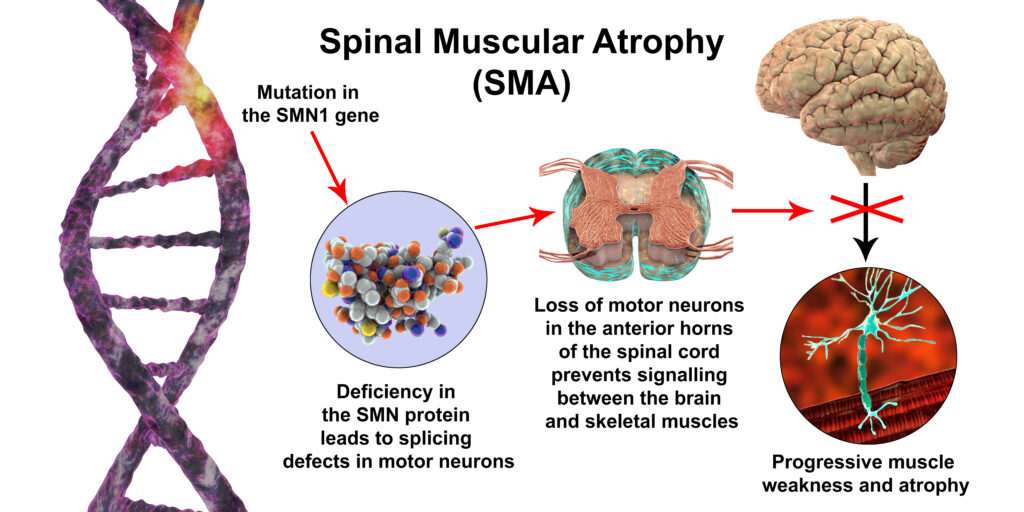
آتروفی عضلانی نخاعی(SMA) یکی از شایع ترین بیماری های نورموسکولار ارثی دوران کودکی است که در اثر نقص در ژن SMN1 ایجاد می شود. این بیماری با از بین رفتن تدریجی نورون های حرکتی نخاعی همراه است و در صورت عدم درمان به موقع، منجر به ضعف پیشرونده عضلات، ناتوانی حرکتی، مشکلات تنفسی و حتی مرگ می شود. با پیشرفت های چشمگیر در ژنتیک پزشکی و درمان های هدفمند، امید تازه ای برای بیماران SMA ایجاد شده است. در این مقاله، علاوه بر علائم و درمان های SMA، به روش های غربالگری و آزمایش های مربوط به تشخیص این بیماری نیز خواهیم پرداخت.
SMA چیست؟
SMA یک اختلال نورودژنراتیو ارثی است که عمدتاً نورونهای حرکتی قدامی در نخاع را درگیر میکند. از نظر بالینی، بیماران دچار ضعف عضلات ارادی، بهویژه عضلات پروگزیمال (نزدیک به تنه) میشوند. این بیماری معمولاً در اثر حذف هوموزیگوتی ژن SMN1 در کروموزوم 5q13 ایجاد میشود.
انواع بالینی SMA
بر اساس سن شروع علائم و سطح عملکرد حرکتی، SMA به چهار زیرگروه بالینی اصلی تقسیم میشود:
- نوع 1 (Werdnig-Hoffmann): شروع در نوزادی؛ شدیدترین نوع؛ نوزاد قادر به نشستن نیست؛ بدون درمان، بقا به زیر 2 سال میرسد.
- نوع 2: شروع بین 6 تا 18 ماهگی؛ بیمار میتواند بنشیند اما قادر به راه رفتن نیست.
- نوع 3 (Kugelberg-Welander): شروع پس از 18 ماهگی؛ بیمار قادر به راه رفتن است اما ضعف عضلانی تدریجی دارد.
- نوع 4: شروع در بزرگسالی؛ فرم خفیف با ضعف اندامهای پروگزیمال.
نکته: طبقهبندیهای دیگر مانند “نوع صفر” (prenatal onset) نیز در موارد بسیار شدید مطرح میشود.
ژنتیک بیماری SMA
در بیش از 95٪ بیماران، حذف هوموزیگوت ژن SMN1 عامل اصلی بیماری است. SMN1 ژنی حیاتی برای بقای نورونهای حرکتی است. ژن SMN2، که شباهت زیادی به SMN1 دارد، بهطور طبیعی نمیتواند عملکرد کامل SMN1 را جایگزین کند زیرا اغلب رونوشت ناقص (ناقص از اگزون 7) تولید میکند.
تعداد نسخههای ژن SMN2 بطور کامل با شدت بیماری مرتبط است:
1 نسخه → معمولاً SMA نوع 1
2 نسخه → نوع 1 یا 2
3 نسخه → نوع 2 یا 3
4 نسخه یا بیشتر → فرمهای خفیفتر
وراثت: SMA بهصورت اتوزومال مغلوب منتقل میشود. ناقلین بدون علامت، یک نسخه معیوب از SMN1 را دارند. خطر تکرار بیماری در هر بارداری برای دو والد ناقل، 25٪ است.
تشخیص ژنتیکی SMA
روش تشخیص استاندارد:
بررسی حذف ژن SMN1 از طریق تستهای ژنتیکی مولکولی (MLPA، qPCR و NGS) با دقت بالای بیش از 95٪ انجام میشود.
آزمایشهای مکمل:
- شمارش نسخههای ژن SMN2 برای پیشبینی شدت بیماری
- بررسی ناقل بودن در خانوادهها
ارزیابیهای بالینی پس از تشخیص اولیه SMA
پس از تشخیص اولیهی SMA، ارزیابیهای بالینی زیر توصیه میشود:
- ارزیابی رشد: اندازهگیری پارامترهای رشد و مقایسه با نمودارهای استاندارد.
- ارزیابی تغذیه و گوارش: بررسی مشکلات تغذیهای، رفلاکس معده-مری (GERD)، خطر آسپیراسیون، وضعیت تغذیهای و زمان صرف غذا. در صورت وجود دیسفاژی یا خطر آسپیراسیون، بررسی امکان قرار دادن لولهی تغذیهای (گاستروستومی) توصیه میشود.
- ارزیابی تنفسی: بررسی اکسیژنسنجی پالس و کاپنوگرافی؛ در صورت لزوم، ارجاع به متخصص ریه با تجربه در SMA.
- ارزیابی خواب: انجام مطالعهی خواب (پلیسومنوگرافی) برای بیماران با مشکلات تنفسی شبانه.
- ارزیابی اسکلتی-عضلانی: بررسی توسط تیم توانبخشی شامل فیزیوتراپی و کاردرمانی برای ارزیابی مهارتهای حرکتی درشت و ظریف، وجود کانترکچرها، دررفتگی مفصل ران، اسکولیوز.
- ارزیابی هماتولوژیک: بررسی ترومبوسیتوپنی و اختلالات انعقادی قبل از تجویز داروها.
- مشاوره ژنتیک: ارجاع به متخصص ژنتیک بالینی یا مشاور ژنتیک برای مشاورهی ژنتیک و بررسی ناقل بودن در خانواده.
- حمایتهای اجتماعی: ارزیابی ساختار خانواده و نیاز به منابع حمایتی مانند گروههای پشتیبانی، مددکاری اجتماعی و پرستاری در منزل.
تشخیص افتراقی SMA
در تشخیص افتراقی SMA باید بیماریهای زیر را در نظر گرفت:
- دیستروفی عضلانی مادرزادی: اختلالات عضلانی با شروع در نوزادی که ممکن است با ضعف عضلانی مشابه SMA باشد.
- میوپاتیهای متابولیک: اختلالات متابولیکی عضلات که میتوانند علائم مشابهی ایجاد کنند.
- نوروپاتیهای ارثی: مانند بیماری شارکو-ماری-توث که با ضعف عضلانی و آتروفی همراه است.
- اختلالات نورون حرکتی دیگر: مانند آمیوتروفیک لترال اسکلروزیس (ALS) در بزرگسالان.
آزمایشهای ژنتیکی مولکولی در SMA
- آزمایش حذف ژن SMN1: بررسی وجود یا عدم وجود ژن SMN1 برای تشخیص SMA.
- شمارش نسخههای ژن SMN2: تعیین تعداد نسخههای ژن SMN2 برای پیشبینی شدت بیماری و پاسخ به درمان.
- آزمایشهای تکمیلی: در مواردی که حذف SMN1 شناسایی نمیشود، بررسی جهشهای نقطهای یا سایر تغییرات ژنی.
درمانهای نوین
در سالهای اخیر، درمانهای مؤثری برای SMA توسعه یافتهاند که بیشتر بر افزایش سطح پروتئین SMN تمرکز دارند:
1. نوزینرسن (Spinraza):
آنتیسنس الیگونوکلئوتیدی تزریقی به مایع نخاعی که اسپلیسینگ ژن SMN2 را اصلاح میکند. اثربخشی بالا در کودکان و حتی بزرگسالان دیده شده است.
2. اوناسمنوژن آبهپارووک (Zolgensma):
ژندرمانی مبتنی بر ویروس AAV9 که نسخه عملکردی ژن SMN1 را به بدن وارد میکند؛ مناسب برای کودکان زیر 2 سال. یکبار تزریق کافی است.
3. ریسدیپلام (Evrysdi):
دارویی خوراکی که مانند نوزینرسن، اسپلیسینگ SMN2 را اصلاح میکند؛ در خانه قابل مصرف است.
مراقبتهای حمایتی
در کنار درمانهای دارویی، حمایتهای چندبخشی شامل فیزیوتراپی، کاردرمانی، تنفسدرمانی، تغذیه مناسب و مراقبت ارتوپدی نقش حیاتی در کیفیت زندگی بیماران دارد.
جمعبندی
SMA یک بیماری ژنتیکی پیشرونده است که تا چند سال پیش بدون درمان مؤثر باقی مانده بود. اما اکنون با پیشرفتهای شگفتانگیز در ژندرمانی و اصلاح اسپلیسینگ، میتوان آیندهای روشنتر برای بیماران ترسیم کرد. تشخیص زودهنگام، آزمایش ناقلین، و مشاوره ژنتیک کلید اصلی مدیریت موفق SMA هستند.